脱リン酸化酵素Ctdnep1 が破骨細胞の分化を抑制する ~骨疾患の新たな治療戦略開発に向けて~
【研究の要旨とポイント】
破骨細胞の過剰な活動は、骨粗鬆症などの骨疾患の原因となることから、分化メカニズムの解明が求められています。
脱リン酸化酵素 Ctdnep1 が、破骨細胞分化のマスター転写因子であるNfatc1のタンパク質レベルを低下させ、破骨細胞の分化を抑制することを明らかにしました。
Ctdnep1によって、破骨細胞分化シグナルであるRANKLのシグナル伝達が抑制されました。
骨疾患の新たな治療戦略の開発につながる知見として期待されます。
【研究の概要】
東京理科大学薬学部生命創薬科学科の早田 匡芳教授、金野 琢人氏(修士課程2年)、村地 眸氏(2021年度薬学科卒業)らのグループは、脱リン酸化酵素の一種である Ctdnep1 が、破骨細胞の分化を抑制することを明らかにしました。
骨の恒常性は、破骨細胞による骨吸収と骨芽細胞による骨形成のバランスによって、維持されています。しかし、過剰な破骨細胞の活動は、骨粗鬆症や関節リウマチ、がんの骨転移による骨破壊などにも関連しており、破骨細胞は骨‧関節疾患の重要な治療ターゲットとなっています。
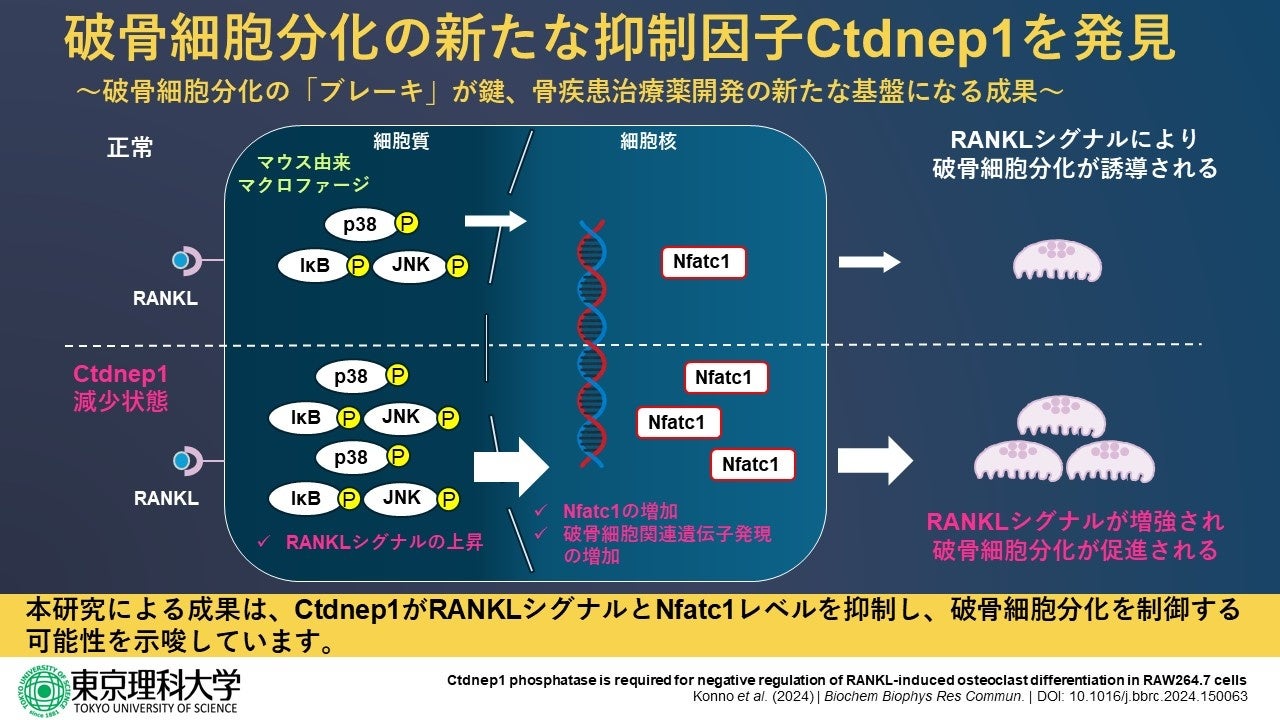
そこで、本研究では、骨格発生、神経発生、腎臓や卵巣の恒常性維持に重要な役割を果たすことが知られている脱リン酸化酵素Ctdnep1に着目し、破骨細胞分化に果たす役割を検討しました。その結果、Ctdnep1は、破骨細胞分化のマスター転写因子(*1)であるNfatc1のタンパク質レベルを低下させ、RANKL(*2)のシグナル伝達を抑制することが明らかになりました。これは、Ctdnep1がRANKLシグナル伝達経路の抑制を通じて破骨細胞の分化を負に制御している(抑制している)ことを示唆しています。
本研究は、リン酸化・脱リン酸化に関わる因子群が破骨細胞の分化制御に果たす役割についての理解を推し進める成果であり、破骨細胞の過剰な活動に関連する骨疾患の新たな治療戦略の開発につながることが期待されます。
本研究成果は、2024年5月7日に国際学術誌「Biochemical and Biophysical Research Communications」にオンライン掲載されました。
【研究の背景】
破骨細胞は、RANKLシグナルによって、免疫系の細胞であるマクロファージが分化してできる細胞です。そのため、RANKLシグナルを遮断する抗体医薬が骨粗鬆症の治療薬として使用されています。しかし、既存薬では十分な効果が見られない患者も存在することから、新たな作用機序でRANKLシグナルを遮断する治療薬の開発が望まれています。
そこで本研究では、骨格発生、神経発生、腎臓や卵巣の恒常性維持に重要な役割を果たすことで知られる脱リン酸化酵素Ctdnep1に着目し、RANKLシグナルによる破骨細胞分化において、どのような役割を果たすか検討しました。
Ctdnep1はBMP(*3)のI型受容体(*4)を脱リン酸化し、II型受容体(*4)を細胞内に取り込むことで、BMPシグナル伝達を阻害します。また、早田教授らは、Ctdnep1を四肢特異的に欠失させると、TGF-β(*5)シグナルが増強され、骨格形成に異常が生じることを過去に報告しています。こうしたことから、Ctdnep1はBMPシグナル・TGF-βシグナルの抑制因子としてはたらくことが示唆されています。
BMP/TGF-βシグナルは、RANKLによる破骨細胞分化を促進するのか、抑制するのか、あるいは関与しないのか未だ統一的な見解は得られておらず、依然として議論の的となっています。Ctdnep1はBMP/TGF-βシグナルを抑制することから、本研究では、マクロファージ様細胞RAW264.7細胞において、Ctdnep1がBMP/TGF-βシグナルを抑制するかどうか、またRANKL誘導破骨細胞分化にどのような役割を果たすかを検討しました。
【研究結果の詳細】
・Ctdnep1 による破骨細胞の分化抑制
Ctdnep1をsiRNAトランスフェクション(*6)によってノックダウンし、RAW264.7細胞を用いて破骨細胞分化誘導実験を行いました。その結果、Ctdnep1をノックダウンすると、RANKLシグナル伝達によって誘導される破骨細胞分化のマスター転写因子であるNfatc1のタンパク質レベルが、RANKL処理なしサンプルおよびRANKL処理24時間後のサンプルで増加しました。このことから、Ctdnep1がRAW264.7細胞における破骨細胞分化の負の調節因子であることを示しています。
なお、このとき、RANKL処理なしおよびRANKL処理24時間後において、Ctdnep1のmRNAレベルに有意な変化は見られなかったことから、Nfatc1タンパク質の上昇は、転写後または翻訳後の制御であると示唆されました。
・Ctdnep1 による骨吸収の抑制
Ctdnep1をノックダウンした破骨細胞は、コントロールの破骨細胞よりもリン酸カルシウム表面を多く吸収し、Ctdnep1が破骨細胞の吸収活性を負に制御していることも確認されました。
・Ctdnep1 による RANKL シグナル伝達の抑制
RANKLシグナル伝達経路におけるCtdnep1のはたらきを調べるために、RANKLシグナル伝達経路の下流で活性化される細胞内シグナル伝達成分の発現およびリン酸化レベルを測定しました。siRNAトランスフェクションしたRAW264.7細胞を6時間血清枯渇させ、その後RANKL刺激を行い、0、5、15、30、60分後に細胞抽出物を調製しました。Ctdnep1ノックダウン細胞では代謝や増殖、運動など、細胞のさまざまな機能に関わるプロテインキナーゼであるp38、JNK、およびIκBのリン酸化レベルが上昇し、上昇が持続していることが示されました。このことは、Ctdnep1がRANKLシグナル伝達を部分的に抑制していることを示唆しています。
なお、Ctdnep1 はBMPシグナルおよびTGF-β シグナルの伝達を調節することが報告されています。加えて、BMP/TGF-βシグナルはRANKL誘導破骨細胞分化に促進的に働くことから、Ctdnep1はBMP/TGF-βシグナルを抑制することにより破骨細胞分化を抑制している可能性があります。そこで本研究では、BMP標的遺伝子であるSmad6、およびTGF-β標的遺伝子である Smad7、Pmepa1 の発現量を測定しました。しかし、Ctdnep1 による発現の促進は見られず、Ctdnep1がRAW264.7細胞におけるBMP/TGF-βシグナル伝達を抑制していないことが示唆されました。これは、Ctdnep1のノックダウンによる破骨細胞分化の亢進にBMP/TGF-βシグナルは関与しない可能性を示唆しています。
以上の結果から、本研究からCtdnep1がRANKLシグナル伝達およびNfatc1タンパク質レベルを負に調節することが明らかになりました。これは、Ctdnep1が過剰な破骨細胞形成を防ぐために必要な因子であることを意味します。
破骨細胞の機能制御は、関節リウマチや骨粗鬆症などの骨疾患の治療戦略の重要な一翼を担っています。Ctdnep1は小児の悪性脳腫瘍である髄芽腫の原因遺伝子としても同定されており、幅広い疾患で重要な因子です。Ctdnep1の新たな機能に関する知見は、骨代謝学を超えて、医学生物学に広く波及する可能性があります。
早田教授は今回の成果について、「Ctdnep1は、私の大学院時代の研究室が発見した遺伝子ですが、それを引き継いで、20年以上研究を継続しています。これまでに、私は共同研究者の皆さんと一緒に、Ctdnep1が、神経発生、骨格発生、腎臓や卵巣の恒常性維持に重要な役割を果たすということを明らかにしてきました。Ctdnep1は、生体に広く分布することから、他にもさまざまな臓器の機能を制御するのではないかと考えており、今回、破骨細胞分化におけるCtdnep1の新たな機能を見出すことができました。この研究結果が、骨代謝学を超えて医学‧生物学の発展に貢献することを期待しています」とコメントしています。
※本研究は、日本学術振興会 科学研究費補助金(JP21H03381, JP22KJ2800)の助成を受けて実施したものです。
【用語】
*1 マスター転写因子
分化する細胞種を決定する転写因子。
*2 RANKL(Receptor activator of nuclear factor-kappa B ligand)
骨細胞や骨芽細胞の表面に発現している膜貫通タンパク質の1種。破骨細胞の前駆細胞の表面に発現する膜貫通タンパク質「RANK」に結合することで破骨細胞分化を誘導する。
*3 BMP
細胞外へ分泌されるタンパク質。BMPシグナルは初期発生や骨格形成に関わる。
*4 BMP I型/Ⅱ型受容体
細胞膜上にあるBMPのセリン・スレオニンキナーゼ型受容体。
*5 TGF-β
細胞の増殖や分化を制御し、細胞死を促すはたらきを持つサイトカイン。
*6 siRNAトランスフェクション
短鎖干渉 RNA(siRNA)を細胞内に導入することで、人工的に mRNA を分解させ、任意の遺伝子をノックダウンする手法。
【論文情報】
雑誌名:Biochemical and Biophysical Research Communications
論文タイトル:Ctdnep1 phosphatase is required for negative regulation of RANKL-induced
osteoclast differentiation in RAW264.7 cells
著者:Takuto Konno, Hitomi Murachi, Kanon Otsuka, Yuta Kimura, Chisato Sampei, Yasuhiro Arasaki, Yukihiro Kohara, Tadayoshi Hayata
DOI:10.1016/j.bbrc.2024.150063
このプレスリリースには、メディア関係者向けの情報があります
メディアユーザー登録を行うと、企業担当者の連絡先や、イベント・記者会見の情報など様々な特記情報を閲覧できます。※内容はプレスリリースにより異なります。
すべての画像