ミトコンドリア病の新たな原因遺伝子を発見
~ 発達遅滞・小頭症・てんかん合併症例の治療法開発へ期待 ~
順天堂大学大学院医学研究科 難病の診断と治療研究センターの八塚由紀子特任助手、岡﨑康司教授と、千葉県こども病院 村山圭部長および埼玉医科大学小児科 大竹明教授らの共同研究グループは、発達遅滞・小頭症・てんかんを併発する日本人のミトコンドリア病*1の新たな原因遺伝子としてNDUFA8遺伝子を同定しました。また、NDUFA8タンパクの減少がミトコンドリア呼吸に必要な呼吸鎖複合体Ⅰ*2全体の欠損を引き起こすことを明らかにしました。本成果は、ミトコンドリア呼吸鎖異常の新たな発生メカニズムを明らかにしたことで、本疾患の病態解明と診断法・治療法の開発につながることが期待されます。本論文はClinical Genetics誌の2020年8月号に掲載される予定です(オンライン版には2020年5月8日掲載)。
本研究成果のポイント
- 日本人ミトコンドリア病症例のゲノム解析を行い、新たな原因遺伝子を同定
- NDUFA8異常が引き起こすミトコンドリア機能破綻のメカニズムを解明
- ミトコンドリア病の病態解明と診断法・治療法の開発に期待
背景
ミトコンドリアの機能低下が原因で発症する疾患を総称してミトコンドリア病と呼びます。新生児期・乳幼児期に発症する重篤なタイプから成人期に顕在化する軽症なタイプまで様々ですが、多くは共通の特徴として筋肉や神経に症状が見られます。
岡﨑教授らの研究グループは十数年にわたり、千葉県こども病院代謝科、埼玉医科大学小児科と共同で、全国から寄せられるミトコンドリア病疑い症例の生化学診断*3と遺伝子診断に取り組んで来ました。ミトコンドリア病の原因遺伝子はミトコンドリアDNAだけでなく、核のDNAにも存在しており、これまでに350を超える原因遺伝子が明らかにされていること、またミトコンドリア病の遺伝形式は様々である(ミトコンドリア遺伝、常染色体顕性遺伝、常染色体潜性遺伝、X連鎖遺伝)ことから、ミトコンドリア病の遺伝子診断は大変複雑なものとなっています。今回の症例のように新規の原因遺伝子を同定した場合は確かに疾患の原因であることを証明しなければならず、多角的な機能解析実験を必要とします。
本研究では、研究グループに寄せられた発達遅滞・小頭症・てんかんを併発する日本人ミトコンドリア病症例を対象に生化学診断とゲノム解析を組み合わせて、新規の原因遺伝子の探索と病態解明を目指しました。
内容
今回、発達遅滞・小頭症・てんかんを併発する日本人ミトコンドリア病症例患者の血液から抽出したゲノムDNAを対象に全エクソーム解析*4を実施しました。その結果、核ゲノム上のNDUFA8遺伝子を新たな原因遺伝子として同定しました。
ミトコンドリアでは、4つの呼吸鎖複合体が酸化還元反応(呼吸鎖複合体Ⅰ-Ⅳ)に沿って電子を運び、ATPを産生する“酸化的リン酸化(OXPHOS)”が行われています。呼吸鎖複合体Ⅰは、45個のサブユニットで構成される最大の呼吸鎖複合体です(図1)。本研究グループがこれまでに蓄積してきたミトコンドリア病の生化学診断データ(症例数650超)によると、ミトコンドリア呼吸鎖に異常が見られた症例のおよそ8割で、この呼吸鎖複合体Ⅰの活性が消失あるいは低下していました。本症例については遺伝子診断に先だって生化学診断が行われ、患者皮膚線維芽細胞のミトコンドリア呼吸鎖複合体Ⅰの酵素活性が正常レベルの約3割に低下していることがわかりました。NDUFA8タンパクはミトコンドリア膜間腔に存在しますが(図1矢印で示す位置)、今回同定された遺伝子変異の部位についてタンパク質の機能解析を行なったところ、NDUFA8タンパクの減少により呼吸鎖複合体全体の形成に異常(欠損)を認め、呼吸鎖複合体Ⅰの他の構成タンパクと結合する上で重要なアミノ酸であることがわかりました(図1右)。
呼吸鎖複合体Ⅰ欠損症の最も一般的な症状としては、大脳基底核や脳幹の病変、呼吸器異常、筋力低下、発育不全、痙攣、高乳酸血症などが挙げられます。本症例の脳MRI所見では脳梁の菲薄化と小脳萎縮を認めました(図2)。
以上の結果から、ミトコンドリア呼吸鎖複合体Ⅰの構成タンパクの1つであるNDUFA8が遺伝子変異によって失われると、呼吸鎖複合体Ⅰのみならず複合体全体の形成にまで影響が及び、患者の細胞ではミトコンドリア活性が失われ、発達遅滞・小頭症・てんかんを引き起こしていることが明らかになりました。
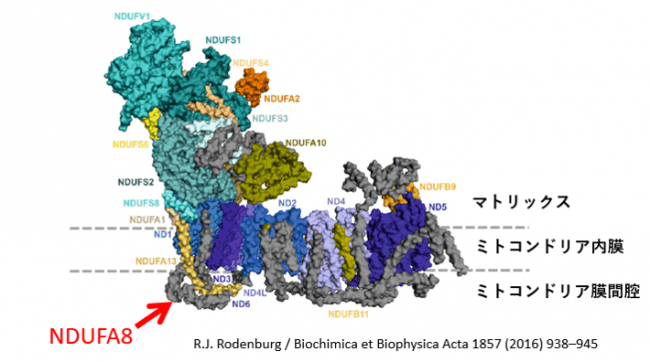
上:ミトコンドリア呼吸鎖複合体ⅠにおけるNDUFA8の位置
下:NDUFA8の変異箇所の構造模式図
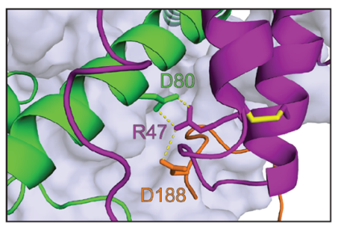
オレンジ: NDUFA13
緑: NDUFB5
NDUFA8タンパクのアルギニン(R47)がシステインに置換される変異が同定された。
R47の側鎖は呼吸鎖複合体Ⅰの形成において他のタンパクとの相互作用に重要である。
図2、脳MRIのT1強調画像*5 (左:3歳時、右:19歳時)
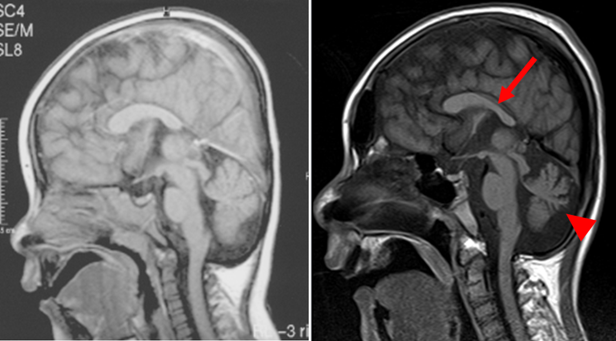
(24歳で本研究グループに紹介)
脳MRI所見では、脳梁が菲薄化し(矢印)、小脳萎縮(矢尻)が認められた。
今後の展開
本成果は、ミトコンドリア病の中で最も多い、ミトコンドリア呼吸鎖複合体Ⅰ活性低下の発生メカニズムの一端を新たに明らかにしたことで、本疾患の病態解明と診断法・治療法の開発につながることが期待されます。
ミトコンドリア病の遺伝子診断は大変複雑で、本研究では全エクソーム解析により新規原因遺伝子の同定に成功しましたが、より多くの患者さんがより早期に遺伝子診断を受けるためには、全エクソーム解析とミトコンドリア病の既知原因遺伝子を包括的に解析するパネル解析*6の両方の整備が必要です。本研究グループで実施中のパネル解析は、現在、保険収載と民間会社への技術移管を目指した開発が進められています。また、症例ごとにより効果的なプラットフォーム選択(全エクソーム解析か、パネル解析)が行えるよう、臨床所見のスコア化と、スコアに応じたプラットフォーム選択基準の整備にも取り組んでいます。
用語解説
*1 ミトコンドリア病: ミトコンドリア病とは、ミトコンドリアの働きが低下することが原因で起こる病気の総称です。エネルギー代謝系(ミトコンドリア呼吸鎖)の先天代謝異常症です。出生5,000人に1人の割合で発症し、いかなる臓器・組織、年齢、遺伝形式でも発病します。特に幼少時期発症例は症状が多様で重篤致死の症例が多いことが知られています。
*2 呼吸鎖複合体Ⅰ: ミトコンドリアでATPを産生する呼吸鎖複合体Ⅰ-Ⅳのうち、 45個のサブユニットで構成される最大の呼吸鎖複合体です。ミトコンドリア病では呼吸鎖複合体Iの異常によるものが多いとされます。
*3 生化学診断: ミトコンドリア病の生化学診断として、ミトコンドリア呼吸鎖の酵素活性や呼吸鎖複合体タンパクの量に異常があるかどうかを調べます。患者皮膚から樹立した皮膚線維芽細胞を使用することが多いですが、生検組織や剖検組織を使用することもあります。
*4 全エクソーム解析: 全エクソーム解析(Whole exome sequencing; WES)は、ゲノムの中でタンパク質をコードするエクソン領域(ヒトゲノムのうち2%未満に相当)を選択的に配列解読する手法です。エクソンは、きわめて重要な領域のため、ここに多くの疾患の原因となる変異が存在しています。
*5 T1強調画像: MRIではT1強調画像とT2強調画像をセットにして撮影し、その組み合わせで組織を特定します。T1強調画像では脂肪や骨髄が白く写り、組織の形状を明瞭に捉えることができます。
*6 パネル解析: 全エクソーム解析が、全遺伝子のエクソン領域を網羅的に配列解読するのに対して、パネル解析では、特定の疾患(ここではミトコンドリア病)の原因遺伝子群に絞ってエクソン領域を調べます。ミトコンドリア病のパネル解析の場合は核にコードされた原因遺伝子およそ350の他、ミトコンドリアDNA全長およそ16kbも対象に含みます。標的領域を絞っているので、全エクソーム解析に比べて配列解析が簡便でコストも低く抑えることが出来ますが、既知の原因遺伝子のみを解析対象としているため新規原因遺伝子を見付けることは出来ません。
原著論文
本研究はClinical Genetics誌の2020年8月号に掲載される予定です(オンライン版には2020年5月8日掲載)。
タイトル: A homozygous variant in NDUFA8 is associated with developmental delay, microcephaly, and epilepsy due to mitochondrial complex I deficiency
タイトル(日本語訳): NDUFA8ホモ接合型変異は、ミトコンドリア複合体I欠損による発達遅滞、小頭症、てんかんに関連する
著者:Yukiko Yatsuka, Yoshihito Kishita, Luke E. Formosa, Masaru Shimura, Fumihito Nozaki, Tatsuya Fujii, Kazuhiro R. Nitta, Akira Ohtake, Kei Murayama, Michael T. Ryan, Yasushi Okazaki
著者(日本語表記):八塚由紀子1)2), 木下善仁1)2), Luke E. Formosa3), 志村優4), 野崎章仁5), 藤井達哉5), 新田和広1)2), 大竹明6)7), 村山圭4), Michael T. Ryan3), 岡﨑康司1)2)8)
著者所属:1)順天堂大学 難病の診断と治療研究センター、2)順天堂大学大学院医学研究科 難治性疾患診断・治療学講座、3)Monash University Department of Biochemistry & Molecular Biology、4)千葉県こども病院 代謝科、5)滋賀県立小児保健医療センター 小児科、6)埼玉医科大学 難病センター、7)埼玉医科大学 小児科学・ゲノム医療学、8)国立研究開発法人 理化学研究所 生命医科学研究センター 応用ゲノム解析技術研究チーム
DOI: 10.1111/cge.13773
本研究は、AMED難治性疾患実用化研究事業(JP20ek0109468, JP19ek0109273)、AMED臨床ゲノム情報統合データベース整備事業(JP19kk0205014)、AMEDゲノム創薬基盤推進研究事業(JP20kk0305015)、JSPS科研費 基盤B(JP19H03624)、 文部科学省 私立大学研究ブランディング事業、豪国The National Health and Medical Research Council(1164459, 1107094)の支援を受け、多施設との共同研究を基に実施されました。
本研究にご協力いただいた皆様に深謝いたします。
このプレスリリースには、メディア関係者向けの情報があります
メディアユーザーログイン既に登録済みの方はこちら
メディアユーザー登録を行うと、企業担当者の連絡先や、イベント・記者会見の情報など様々な特記情報を閲覧できます。※内容はプレスリリースにより異なります。
すべての画像
